


Sickle Cell Disease And The Era Of CRISPR

I first heard about sickle cell while watching ESPN. The anchor had just reported that Ryan Clark, a Pittsburgh Steelers safety, would not be playing in Denver that weekend. Several years earlier, after also playing in Denver, Clark needed surgery on his gallbladder and part of his spleen due to complications from his sickle cell trait. Sickle cell trait is a condition affecting between 1 and 3 million Americans and more than 100 million people worldwide, caused by inheriting one normal gene and one sickle gene. Clark’s situation was uncommon, as most people with sickle cell trait have no symptoms.
In contrast, a person who inherits two sickle genes has sickle cell disease (SCD), a more serious genetic disorder which afflicts 100,000 Americans and 20 million people worldwide. SCD causes flawed hemoglobin, the protein in red blood cells which delivers oxygen to tissues in the body. When oxygen leaves the cells, they become sickle-shaped and get stuck together, leading to blood vessel blockages, pain, and anemia. These cells have a lifespan that is just one-tenth that of normal cells.
 | CDC")
In recent years, awareness of the danger of SCD has risen immensely. A 2023 study found that SCD could be responsible for eleven times more deaths than previously reported. This is because SCD increases the risk of infection and death from other health issues like heart problems and kidney disease. Instead of the previously estimated 34,000 cause-specific sickle cell deaths, the “total mortality burden”—including deaths from related complications—was 373,000 people. This places total SCD as the 12th leading cause of death in children, compared to 40th for cause-specific SCD. In sub-Saharan Africa, home to around 70-80% of SCD cases, it is the 11th leading cause of death in children.
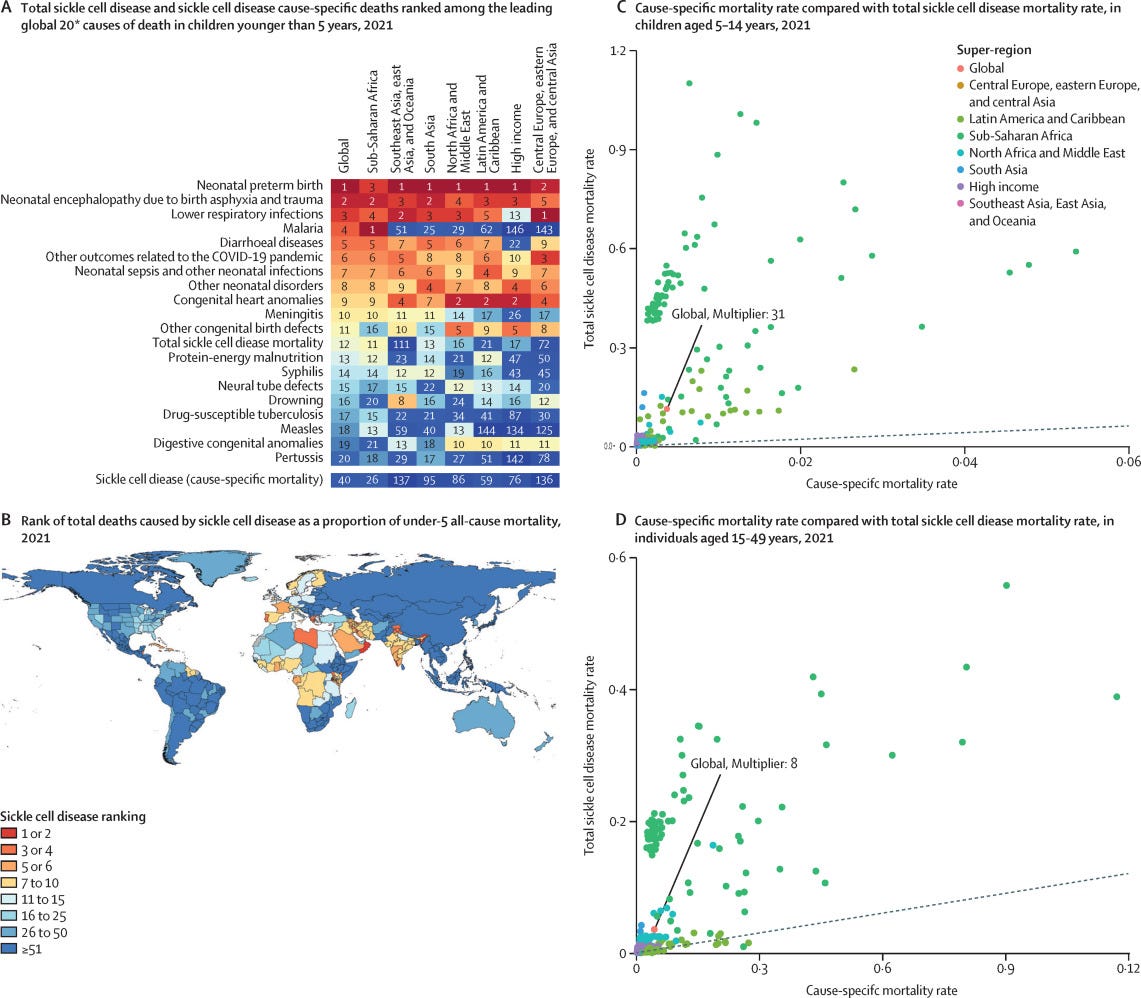
In the United States, more than 90% of SCD patients are Black—with the disease affecting one in every 365 Black people. Another 3-9% of patients are Hispanic or Latino. The lifespan of those with SCD is more than 20 years shorter than average expected life expectancy.
The first cure for SCD came in 1984, with the first successful bone marrow transplant. While miraculous, bone marrow transplants have serious drawbacks. The procedure requires a full donor match—only 15-30% of people have a full match in their family—and comes with various risks, including, in rare cases, death.
In the past forty years, four treatments have also been approved for SCD. The first was Hydroxyurea, an oral medicine that decreases sickling. It is generally the standard treatment applied, with the other treatments used as an additive if it cannot reduce symptoms sufficiently. The other drugs aid in managing different parts of SCD, including stopping sickling further, lowering the number of pain crises, and preventing the blockage of blood flow. These treatments have all proven their efficacy, but last year saw the emergence of new treatments that promise greater hope in the work to end SCD.
On December 8, 2023, the FDA approved two treatments, Casgevy and Lyfgenia, for SCD. Lyfgenia involves inserting a gene into red blood cells that allows them to produce normal hemoglobin, thus preventing sickling. 88% of those in a Lyfgenia trial had no pain six to eighteen months after the gene insertion. Several weeks ago, the first patient, a twelve-year old boy in Washington, DC, began treatment with Lyfgenia. It will take months for the treatment to occur, and at present, only a few dozen patients will start treatment this year. However, tens of qualified treatment centers are already accepting patients with an eye to scaling up in the future.
The other treatment, Casgevy, is the first therapy approved in the US that uses CRISPR. CRISPR is a revolutionary gene editing technology which allows scientists to edit the DNA of living beings. In this case, a patient’s cells are collected, then modified using the technology to prevent red blood cell sickling, and then transplanted back into the patient. In a trial, 97% of patients who previously experienced multiple severe pain crises a year went at least a year without severe pain in the two years after receiving the treatment.

If CRISPR were to play a role in ending SCD, that alone would be an astonishing success. But we might just be scratching the surface of its potential impact. Casgevy has also been approved for beta-thalassemia, another severe genetic blood disorder. Those with beta-thalassemia major have a life expectancy of 17 years, and few live beyond 30 years old. An ongoing trial is also investigating the possibility of using CRISPR to lower cholesterol. As well, there are clinical trials using CRISPR to tackle cancer, cardiovascular disease, HIV/AIDS, UTIs, diabetes, and more.
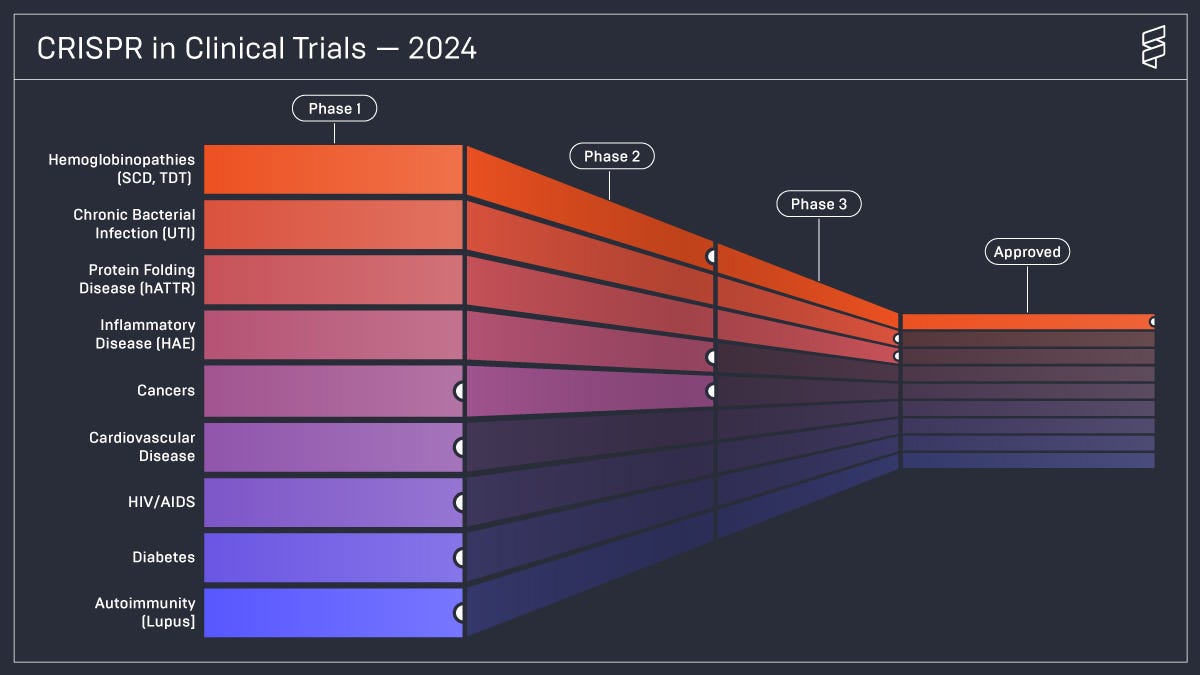
While CRISPR shows extraordinary promise, the end to SCD is far from assured. Casgevy—as well as Lyfgenia—is incredibly expensive, meaning that a majority of those who need the treatment do not have access to it. The Biden administration has announced an effort to allow the Centers for Medicare and Medicaid Services to negotiate discounts with the purveyors of both treatments, with the goal of launching in 2025.
In addition, the ability to edit DNA raises ethical concerns, with fears of scientists attempting to play god. In at least one famous circumstance, these fears were well-founded, as scientists in China created “CRISPR babies”, using the technology to edit DNA in embryos. However, since that incident, a growing consensus has emerged against using CRISPR to edit inherited genes, while allowing for changes in blood, muscle, or brain cells.
In spite of these very real concerns, CRISPR is an extraordinary accomplishment. It was only twelve years ago that scientists discovered CRISPR-Cas9, the specific technology being used in Casgevy. In a little over a decade, we have gone from a theoretical discussion around gene-editing to this technology being applied in the real world to treat deadly, painful diseases. It is particularly important, in my mind, that the first application of this groundbreaking technology is tackling a disorder that disproportionately affects people who have been marginalized in health care systems and by medical communities for far too long, both in the US and globally.
CRISPR’s demonstrated success in treating SCD should already give us hope, as it might rid a great number of people of incredible pain and improve their quality of life. As scientists and policymakers work to expand access to these extraordinary treatments for SCD, we will also see if CRISPR has the potential to change the world even further. It took twelve years from the discovery of CRISPR-Cas9 to get to this point. We can only begin to imagine the incredible progress that will follow in the next twelve.